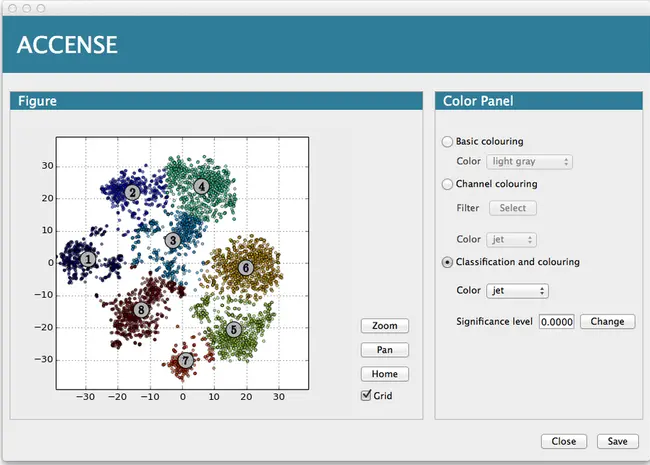
ACCENSE is a tool for exploratory analysis of high-dimensional single-cell data such as that generated by Mass Cytometry. By combining a nonlinear dimensionality reduction algorithm with a k-means clustering algorithm both visualization for exploratory analysis and automated cell classification into subpopulations is performed.
By automating cell classification, yet retaining single-cell resolution, ACCENSE facilitates exploratory data analysis while circumventing the need for any manual "gating". Importantly, by outputting tabular single-cell data, ACCENSE facilitates downstream statistical analysis.
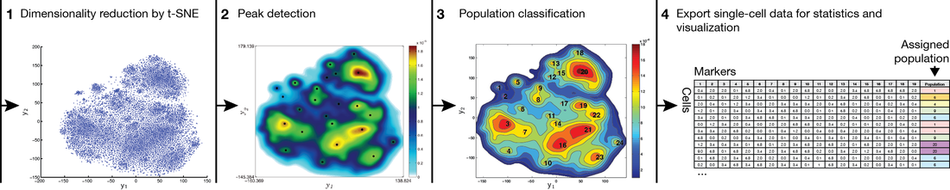
The dimensionality reduction step within ACCENSE employs t-distributed Stochastic Neighbor Embedding (t-SNE), published in van der Maaten L and Hinton G, Journal of Machine Learning Research, 9 (2008) 2579-2605 or a Barnes-Hut SNE algorithm published in van der Maaten, Proceedings of the International Conference on Learning Representations, 2013.
ACCENSE was developed by Petter Brodin (Karolinska Institutet and previously Mark Davis lab at Stanford University), and Karthik Shekhar (prof. Arup Chakraborty at M.I.T. and the Ragon Institute of MIT, MGH and Harvard).
ACCENSE is a standalone application for exploratory analysis of high-dimensional single-cell data such as that generated by Mass Cytometry. The main functions that ACCENSE provides are:
- It performs a nonlinear dimensionality reduction on the high-dimensional single-cell data and obtains the inferred low-dimensional representation.
- The low dimensional data can be visualized with abundant coloring options.
- It provides clustering methods to automate the classification of cellular sub-populations.
ACCENSE is developed based on the algorithm proposed in, with improved classification methods. It is written in Python and employs the following third-party packages.
The current version above has been developed and tested in Mac OSX Mavericks (10.9). Support for additional Mac OS and Windows will be added shortly.
The dimensionality reduction step within ACCENSE employs t-distributed Stochastic Neighbor Embedding (t-SNE), published in van der Maaten L and Hinton G, Journal of Machine Learning Research, 9 (2008) 2579-2605 or a Barnes-Hut SNE algorithm published in van der Maaten, Proceedings of the International Conference on Learning Representations, 2013.


Unlock the power of high-dimensional single-cell data with our cutting-edge tools designed for exploratory analysis. Our platform provides researchers, bioinformaticians, and data scientists with intuitive and robust solutions to delve into the intricate world of cellular diversity and function.
Why Choose Our Tools?

Comprehensive Analysis
Explore complex single-cell datasets with advanced dimensionality reduction, clustering, and visualization techniques.

User-Friendly Interface
Seamless integration for researchers with or without coding expertise. Our intuitive tools ensure accessibility for all levels of experience.
Customizable Workflows
Tailor your analysis pipeline to meet the unique demands of your research.

Scalable Performance
Handle datasets of any size, from small pilot studies to large-scale projects, without compromising speed or accuracy.
Features
Dimensionality Reduction
James Mitchell is an esteemed IT university professor with over 20 years of experience in teaching and research.
Clustering and Classification
Identify distinct cell populations and classify them based on their unique signatures.
Interactive Visualization
Visualize your data in 2D and 3D plots, heatmaps, and custom dashboards.
Data Integration
Combine multi-modal single-cell data for a holistic view of cellular function.
Statistical Insights
Gain deeper biological insights with integrated statistical and machine learning tools.